Biosimilars put biologic medicines within reach for more patients
Making life-changing therapies more affordable
With demonstrated biosimilarity to the reference products at lower costs to patients and providers, biosimilars play an important role in the future of health care by improving access to medications and encouraging investments in research and innovation. Biosimilars can potentially offer up to 30% price reductions when compared with reference products.1 These savings allow physicians to follow established treatment guidelines without compromises based on cost. Thanks to biosimilars, direct spending on biologic drugs in the U.S. is estimated to drop by $54 billion between 2017 and 2026. 2The High Cost of Biologics3

The High Cost of Biologics3
% of Biologics used in the U.S.

% of prescription drug spending on Biologics

%
The BioSpecialized Journey
What it takes to make life-changing medicines

Our biosimilar process

Regulatory Approval

Interchangeability & Switching
What does it take to make life-changing medicines?

What does it take to make life-changing medicines?
- Genetic instructions are inserted into a clone (cell) to produce a specific candidate biologic3
- After different versions of the biologic are made, each one by a different cell, screening is conducted to identify the cell line that produces the protein that is most similar to the reference product3
- A process to manufacture the biosimilar is developed, in a way that maximizes the similarity between the biosimilar and the reference product3,4
Characterize Reference Product
- Assess variability to identify target range
- Understand critical quality attributes or reference product (immunology, safety/toxicity, PK/PD, efficacy)
Target-directed Iterative Development
- Develop unique cell line and manufacturing process
Establish Similarity
- Physicochemical, biologic and functional characterization
- Confirm critical quality attributes are similar to reference product
Additional Information to Supplement Similarity
- Non-clinical, PK/PD studies
- Clinical trials to confirm comparability (safety, efficacy, immunogenicity) and eliminate any residual uncertainty
PD: pharmacodynamics; PK: pharmacokinetics
Development of Fresenius Kabi biosimilars
Equipped with global expertise in biologic development and manufacturing, Fresenius Kabi applies the same high quality standards to developing and producing biosimilars that are required for the development of reference products. We bring years of pharmaceutical experience and technical knowledge to ensure the production of a consistent, high-quality product. Fresenius Kabi biosimilars meet the high regulatory standards required by the FDA, based on totality of evidence across analytical, pharmacokinetic/pharmacodynamic (PK/PD), clinical safety and immunogenicity assessments against the reference product.6- EMA. http://www.ema.europa.eu/docs/en_GB/document_library/Leaflet/2017/05/WC500226648.pdf. Published 2017. Revised 2019. Accessed May 5, 2021.
- FDA. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/quality-considerations-demonstrating-biosimilarity-therapeutic-protein-product-reference-product. Published 2015. Accessed May 5, 2021.
- Gupta SK, et al. http://www.bioprocessintl.com/manufacturing/biosimilars/opportunities-challenges-biosimilar-development. Published 2017. Accessed May 5, 2021.
- Al-Sabbagh A, et al. Development of biosimilars. Semin Arthritis Rheum. 2016; 45(5 suppl):S11–S18.
- mAbxience. https://www.mabxience.com/products/biosimilar/generics-biologics-biosimilars-whos-who. Revised 2021. Accessed May 5, 2021.
- GaBi. https://www.gabionline.net/Biosimilars/News/Fresenius-Kabi-s-pegfilgrastim-biosimilar-accepted-for-review-by-EMA-and-FDA. Published 2020. Accessed May 5, 2021.
Our biosimilar process

Our biosimilar process
- The chosen cell line is expanded in large bioreactors, in conditions optimized for protein production2,3
- The synthesized biologic is isolated and purified, removing any unwanted impurities2,3
- The harvested protein is analyzed for uniformity in its 3D structure and potency2
Cell line expansion in bioreactors
Protein retrieved via centrifugation
Purification with chromatography
Analysis for uniformity
- Kabir ER, et al. The breakthrough of biosimilars: A twist in the narrative of biological therapy. Biomolecules. 2019;9(9):410.
- Gupta SK, et al. http://www.bioprocessintl.com/manufacturing/biosimilars/opportunities-challenges-biosimilar-development. Published 2017. Accessed May 5, 2021.
- Al-Sabbagh A, et al. Development of biosimilars. Semin Arthritis Rheum. 2016; 45(5 suppl):S11–S18.
Regulatory Approval

Regulatory Approval
The FDA regulatory approval process has been designed to robustly assess each biosimilar submitted on a case-by-case basis to ensure that health care providers and patients can rely on their efficacy, quality and safety.1
The licensure pathway
Due to the complexity of the manufacturing process of biologics, the small molecule drug approval process cannot be applied to biosimilars. Instead, biosimilars undergo an abbreviated licensure pathway to which the FDA still applies the same high standards of safety and efficacy as it does for reference biologics or small molecule drugs.2
While clinical trials for reference biologics are designed to demonstrate superiority, the goal of biosimilar clinical trials is to provide proof of quality and similarity in safety and efficacy. Studies to evaluate mechanism of action, determine dosing, or show patient benefit are not needed for biosimilars, because these were all established by the reference product.1,3,4
This licensure pathway may result in faster access to biosimilars, providing health care providers and patients with additional therapeutic options while maintaining confidence in their efficacy, quality and safety.1
The totality of evidence approach
The FDA applies the ‘totality of evidence’ approach to biosimilar regulatory approval, requiring a rigorous stepwise process of comparison with the reference product.1,2Assessing the ‘totality of evidence’ involves thorough examination of all data submitted from extensive analytical, non-clinical and comparative clinical studies.1
To perform any comparator studies, a complete understanding of the relationship between structure and function of the reference product must be obtained.5 Data is analyzed and evaluated after each stage to identify any residual uncertainty about biosimilarity and eliminate it through further studies.1
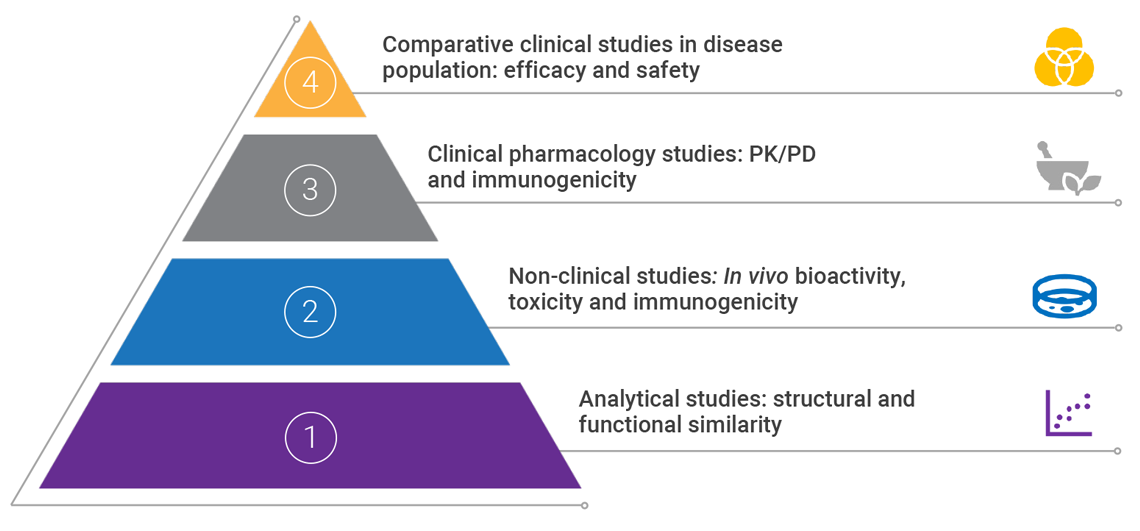
PD: pharmacodynamics; PK: pharmacokinetics
Adapted from Janjigian YY, et al 2018 and Markus R, et al. 2017.2,5
Demonstrating biosimilarity
An application for the approval of a biosimilar or interchangeable product must include data demonstrating biosimilarity to the reference product. This usually includes data from:- Analytical studies: demonstrating that the biologic is highly similar to the reference product, in spite of minor differences in clinically inactive components6
- Non-clinical studies, including an assessment of toxicity6
- A clinical study (or studies) that demonstrates safety, purity and potency of the proposed biosimilar product in one or more indications for which the reference product is licensed. This usually includes assessing immunogenicity, pharmacokinetics, and, in some cases, pharmacodynamics, and may also include a comparative clinical study6
- The proposed interchangeable product is expected to produce the same clinical result as the reference product in any patient6
- For a product administered more than once to a patient, switching between the proposed interchangeable product and the reference product does not lead to increased safety risks or decreased effectiveness compared to using the reference product without any switching between products6
The exact FDA data requirements vary with each application depending on:
- The information available for the reference product
- The strength of biosimilarity demonstrated by each round of evidence
Post-marketing safety monitoring
The FDA places strong emphasis on ensuring high quality of manufacture, processing, packaging and storage.1 Heterogeneity is inherent to all biologic drugs and is not limited to biosimilars.2,3 To combat this, the FDA requires extensive characterization of lot-to-lot variability of both reference product and biosimilar. The degree of batch variability must be maintained from the reference product.1 Following approval, biosimilars must undergo the same ongoing safety monitoring as biologics. Evidence from both the regulatory assessment and post-marketing surveillance has confirmed that biosimilars have comparable safety and efficacy profiles to their reference products.2,7-10- FDA. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product. Published 2015. Accessed May 5, 2021.
- Janjigian YY, et al. Talking to patients about biosimilars. Future Oncol. 2018;14(23):2403-2414.
- Declerck P, et al. The language of biosimilars: Clarification, definitions, and regulatory aspects. Drugs. 2017;77(6):671-677.
- Thill M, et al. Biosimilars: what the oncologist should know. Future Oncol. 2019;15(10):1147-1165.
- Markus R, et al. Developing the totality of evidence for biosimilars: Regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017;31(3):175-187.
- U.S. Food and Drug Administration. Biosimilars. 2018. Available at:
https://www.fda.gov/drugs/developmentapprovalprocess/howdrugsaredevelopedandapproved/approvalapplications/therapeuticbiologicapplications/biosimilars/default.htm
(Accessed February 2019). - Braun J, et al. Switching to biosimilar infliximab (CT-P13): Evidence of clinical safety, effectiveness and impact on public health. Biologicals. 2016;44(4):257-266.
- Dörner T, et al. The changing landscape of biosimilars in rheumatology. Ann Rheum Dis. 2016;75(6):974-982.
- Jahnsen J. Clinical experience with infliximab biosimilar remsima (CT-P13) in inflammatory bowel disease patients. Therap Adv Gastroenterol. 2016;9(3):322-329.
- Jørgensen KK, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304-2316.
Interchangeability & Switching

Interchangeability & Switching
‘Switching’ and ‘automatic substitution’ are often mentioned in the context of changing a patient’s prescription from a biologic to a biosimilar. The main difference between the two is that ‘switching’ must be either instigated or approved by the prescriber whereas ‘automatic substitution’ can theoretically be done without the knowledge of the prescriber if the product has been designated as interchangeable by the FDA.1,2
Switching
This is the decision by the treating physician to switch patients from a biologic to a biosimilar, or vice versa. Unless the biosimilar is designated as interchangeable, switching must always be approved by the treating physician.1
Automatic Substitution
A pharmacist substitutes a prescribed product with another interchangeable product without consulting the prescriber.2
Interchangeable biosimilars in the U.S.
To achieve this status, additional studies are needed to demonstrate that:3-5
- The biosimilar produces the same result as the reference product in any patient, and
- Any type of switching of products does not result in safety risk or diminished efficacy compared to the reference product
- Barbier L et al. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: A systematic review. Clin Pharmacol Ther. 2020;108(4):734‒755.
- Declerck P et al. The language of biosimilars: Clarification, definitions, and regulatory aspects. Drugs. 2017;77(6):671‒677.
- Dalpoas SE et al. Barriers to biosimilar utilization in the United States. Am J Health-Syst Pharm. 2020;77(23):2006‒2014.
- Alvarez DF et al. Interchangeability of biosimilars: What level of clinical evidence is needed to support the interchangeability designation in the United States? BioDrugs. 2020;34(6):723‒732.
- FDA. https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval#process. Published 2015. Accessed May 5, 2021.
Frequently asked questions about biosimilars

Biosimilars, like their reference products, are used in many different therapy areas including cancer and a number of autoimmune diseases. Fresenius Kabi is currently developing biosimilar medicines for use in oncology and immunology.
Patients who are already receiving a different biologic treatment can also be changed or ‘switched’ to a biosimilar
Molecular differences
Generics are relatively small, simple molecules that are derived from inorganic sources and manufactured to be exact replicas of the original drug. By contrast, biosimilars are large and complex molecules, often proteins, that are derived from living sources.11Cost of biosimilars vs. generic drugs
The chemical structure of a generic is identical to the original drug. This means that less testing is required for approval, making them around 80% cheaper than the reference product.12,13 The development timeline for a generic drug is typically only around 2-3 years.14Key differences between biosimilar and generic drugs11,12,14-16,18-20
| Biosimilars | Generics | |
|---|---|---|
| Molecular type | Large, complex | Relatively small, simple |
| Molecular source | Living | Inorganic |
| Required to prove | Highly similar rather than identical to reference product. Non-clinical and clinical studies required to demonstrate biosimilarity with the reference product, but not required to independently establish safety and efficacy | Bioequivalent and identical to reference product (in strength, dose and route of administration). No non-clinical or clinical studies required. |
| Immunogenicity testing | Yes | No |
| Nomenclature | Trade names or distinguished reference product established name + 4-letter suffix | International nonproprietary name (INN) |
| Risk management plan | Yes | No |
| Price discount | 20% - 30% discount over the reference product | 80% - 85% discount over reference product |
| Time to market | 7-8 years | 2-3 years |
To learn more, visit: https://www.fda.gov/drugs/therapeutic-biologics-applications-bla/biosimilars

- Kabir ER, Moreino SS, Siam MKS. The breakthrough of biosimilars: A twist In the narrative of biological therapy. Biomolecules. 2019;9{9):410. https://pubmed.ncbi.nlm.nih.gov/31450637/
- lnserro A. Enzi, Hassan introduce bipartisan bill to improve biosimilar education. American Journal of Managed Care (AJMC). Published May 27, 2019. Accessed May 20, 2022. https://www.centerforbiosimilars.com/view/enzi-hassan-introduce-bipartisan-bill-to-improve-biosimilar-education
- Mulcahy AW, Hlávka JP, Case SR. Rand Corporation. Biosimilar Cost Savings in the United States; Initial Experience and Future Potential. https://www.rand.org/content/dam/rand/pubs/perspectives/PE200/PE264/RAND_PE264.pdf. Published 2017. Accessed September 14, 2022.
- Declerck P, et al. The language of biosimilars: Clarification, definitions, and regulatory aspects. Drugs. 2017;77(6):671-677.
- Kabir ER, et al. The breakthrough of biosimilars: A twist in the narrative of biological therapy. Biomolecules. 2019;9(9):410.
- Janjigian YY, et al. Talking to patients about biosimilars. Future Oncol. 2018;14(23):2403-2414.
- Barbier L, et al. The efficacy, safety, and immunogenicity of switching between reference biopharmaceuticals and biosimilars: A systematic review. Clin Pharmacol Ther. 2020;108(4):734-755.
- FDA. https://www.fda.gov/media/82647/download. Published 2015. Accessed May 5, 2021.
- U.S. Food and Drug Administration (FDA). Biosimilar and Interchangeable Biologics: More Treatment Choices, https://www.fda.gov/consumers/consumer-updates/biosimilar-and-interchangeable-biologics-more-treatment-choices#. Updated October 12, 2021. Accessed Aug. 26, 2022.
- FDA. http://www.fda.gov/media/154995/download. Published 2017. Accessed May 5, 2021.
- Thill M, et al. Biosimilars: what the oncologist should know. Future Oncol. 2019;15(10):1147-1165.
- FDA. https://www.fda.gov/media/83670/download. Accessed May 5, 2021.
- CTCA. https://www.cancercenter.com/community/blog/2018/12/whats-the-difference-biosimilar-and-generic-drugs. Published 2018. Accessed May 5, 2021.
- mAbxience. https://www.mabxience.com/products/biosimilar/generics-biologics-biosimilars-whos-who. Revised 2021. Accessed May 5, 2021.
- Bhatt V. Current market and regulatory landscape of biosimilars. Am J Manag Care. 2018;24(21 Suppl):S451‒S456.
- Janjigian YY, et al. Talking to patients about biosimilars. Future Oncol. 2018;14(23):2403-2414.
- FDA. https://www.fda.gov/consumers/consumer-updates/generic-drugs-undergo-rigorous-fda-scrutiny. Published 2014. Accessed May 5, 2021.
- Halimi V, et al. Clinical and regulatory concerns of biosimilars: A review of literature. Int J Environ Res Public Health. 2020;17(16):5800.
- US Pharmacist. https://www.uspharmacist.com/article/biosimilars-not-simply-generics. Published 2019. Accessed May 5, 2021.
- FDA. https://www.fda.gov/drugs/biosimilars/biosimilar-development-review-and-approval. Published 2017. Accessed June 10, 2021.
- Inserro A. https://www.centerforbiosimilars.com/view/enzi-hassan-introduce-bipartisan-bill-to-improve-biosimilar-education. Published 2019. Accessed May 5, 2021.
- Dean EB, et al. Physician, practice, and patient characteristics associated with biosimilar use in medicare recipients. JAMA Netw Open. 2021;4(1):e2034776.
- Mulcahy AW, Hlávka JP, Case SR. Rand Corporation. Biosimilar Cost Savings in the United States; Initial Experience and Future Potential. https://www.rand.org/content/dam/rand/pubs/perspectives/PE200/PE264/RAND_PE264.pdf. Published 2017. Accessed September 14, 2022.
- Dutta B, et al. Identifying key benefits in European off-patent biologics and biosimilar markets: It is not only about price! Biodrugs. 2020;34(2):159-170.
- McKinnon R, et al. Safety considerations of biosimilars. Aust Prescr. 2016;39(6):188-189.